Skip to main content
Search
Search This Blog
Jermdemo
Mostly bioinformatics, NGS, and cat litter box reviews
Posts
Showing posts from 2009
Show all
November 19, 2009
Using Vmatch to combine assemblies
November 04, 2009
R's xtabs for total weighted read coverage
October 23, 2009
Installing Bio::DB::Sam from CPAN
October 12, 2009
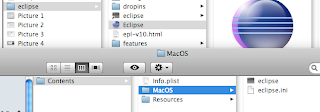
Disable file locking in Eclipse for OS X
September 23, 2009
My 2009 Bridge-to-Bridge Experience
August 26, 2009
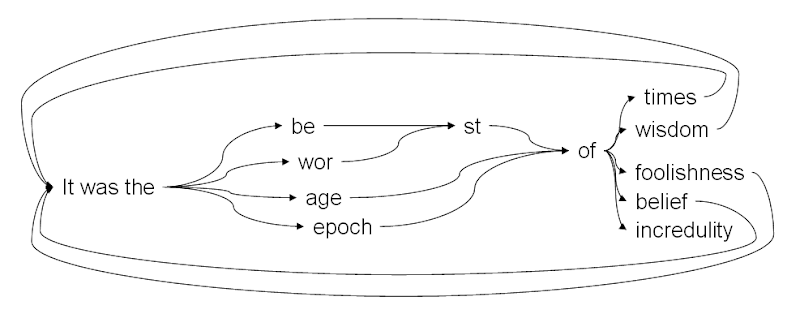
Charles Dickens de Bruijn Graph
August 25, 2009
Standardized Velvet Assembly Report
April 01, 2009
I pick on the WSJ again: A true cost analysis of home prices
February 26, 2009
The WSJ made an accounting error
Newer Posts
Older Posts
Home